A World Model of Protein Biology
Four billion years of evolution have produced the molecular machinery of life: proteins. They form cellular structures, transmit signals, defend against infection, repair damage, and regulate nearly every process that keeps organisms alive. But even as protein structure prediction has advanced, the vast majority of proteins remain unstudied, their functions a mystery.
Today, at Biohub, we are taking a step toward changing that.
We are releasing a world model of protein biology: a scientific engine for prediction, design, and discovery. The next generation of Evolutionary Scale Models (ESM), this system learns from the protein sequences produced by evolution and uses that knowledge to represent, map, predict, and design proteins.
The system includes three artifacts: ESMFold2, a state-of-the-art model that predicts protein structure and powers the design of new proteins; ESM Atlas, a map of 6.8 billion sequences and 1.1 billion predicted structures; and ESMC, a state-of-the-art protein language model trained on approximately 2.8 billion sequences drawn from across all of life.
The results we are sharing today mark significant progress in one of biochemistry's hardest problems: designing proteins that bind to specific molecular targets. We show that ESMFold2 can design high-affinity protein binders — including single-chain antibodies — validated in the lab against five clinically relevant targets in oncology and immunology. Read the full results and methods in our preprint.
We believe this is an important moment in protein design. When digital representations of biology become accurate enough, protein designs can be tested computationally before they reach the bench.
ESMC provides a foundation for modeling the sequence, structure, and function of proteins. ESMFold2 predicts the structure of proteins and biomolecular complexes with state-of-the-art accuracy and speed. Features derived from model representations capture fundamental principles of structure and function, forming a compositional grammar for protein biology.
The next generation of ESM
ESMC is the latest model in a research program that developed the first transformer language model of protein sequences in 2019. The core scientific hypothesis of ESM is that training a language model across the sequences of all life will cause it to internalize the fundamental properties that govern protein biology — the rules underlying how proteins fold, interact, and function.
Proteins are chains of amino acids: 20 chemical building blocks that, when strung together, can form a vast number of combinations. The order of those amino acids determines how the chain folds into a three-dimensional structure, and folding determines function.
We trained the first ESM language models on millions of protein sequences drawn from across life, with a simple objective: predict the amino acids that have been randomly masked out, based on the context of the rest of the sequence. In early research we found what the models learned went beyond sequences. Without any structural supervision, their internal representations encoded biological structure and function, including properties the model had never been explicitly shown.
We also found that what the models learn scales predictably with compute: more training produces richer representations of biology. We extended this work with ESM2, and later ESM3.
ESMC (short for ESM Cambrian) takes this scaling to the next level, training on billions rather than millions of sequences. We identified a scaling law linking the amount of compute power used to train the models and the degree to which their representations accurately capture biology. In ESMC, this powers linear returns with scale, leading to a new state of the art in protein representations.
The internal representations that a language model learns become a powerful substrate for artificial intelligence, predicting structure and function, and generating new sequences.
Structure prediction with language models
A model that has internalized the rules governing protein biology should be able to predict the folded structure of proteins. ESMFold2 passes that test, achieving state-of-the-art accuracy across a broad range of benchmarks, with particularly strong performance on antibody-antigen prediction, one of the hardest and most therapeutically relevant challenges for structure prediction.
ESMFold2 uses a looped transformer architecture. Rather than passing representations through a network once, ESMFold2 loops them through the same parameters multiple times and is optimized through this loop during training. The model learns to use each pass to refine its structural representation based on what it computed in the previous one.
This has two practical consequences. First, ESMFold2 can scale compute at inference time by running more loops, allocating more capacity to harder predictions without retraining. Second, because this depth comes from recurrence rather than additional parameters, the model avoids the overfitting that would otherwise constrain a model trained on the relatively limited pool of experimentally determined protein structures. Taking gradients through the recurrent process during training is what distinguishes ESMFold2's architecture from other structure prediction models that use iterative refinement.
Where most structure prediction models build their representations by searching for evolutionarily related sequences (multiple sequence alignments, or MSAs), ESMFold2 operates directly from ESMC's learned protein representations, capturing evolutionary information encoded during language model pretraining rather than requiring explicit alignment. MSA inputs are supported optionally and improve accuracy when available.
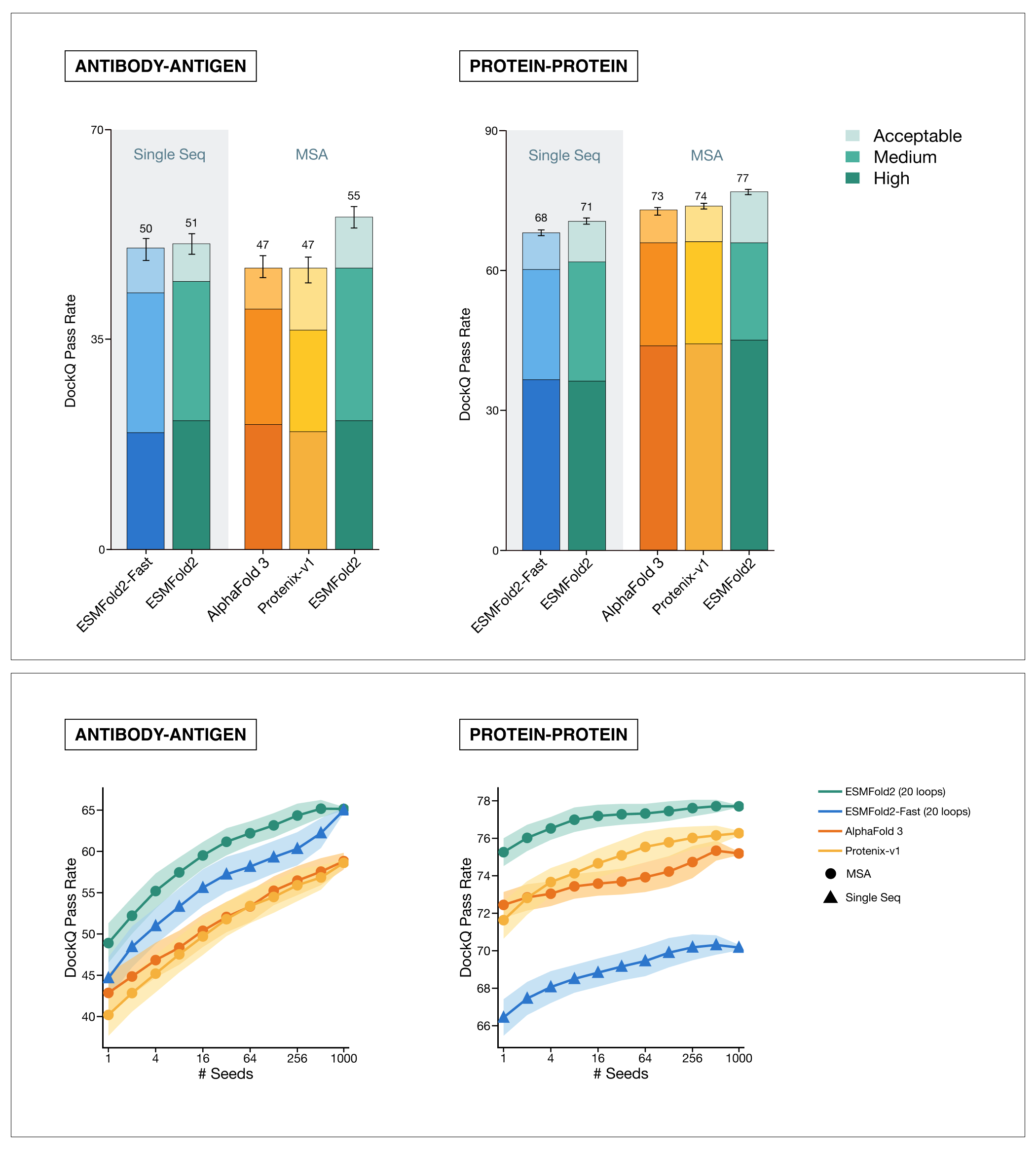
ESMFold2 achieves state-of-the-art accuracy in structure prediction, both for general protein-protein interactions and for the challenging and therapeutically relevant task of antibody-antigen prediction. From ESMC representations alone, ESMFold2 is more successful at predicting the true binding pose of antibody-antigen complexes than AlphaFold 3. When provided with the same evolutionary information (MSA) as AlphaFold, ESMFold2 is the strongest predictor on both benchmarks. Bottom: structure prediction models can benefit from a larger computational budget. When we let models make multiple predictions and score them based on their own confidence estimates, ESMFold2 consistently improves with more compute.
ESMFold2 achieves state-of-the-art accuracy on Foldbench, a suite of structure prediction benchmarks, correctly predicting 55% of antibody-antigen complexes. For protein-protein interactions more broadly, ESMFold2 correctly predicts 71% of complexes from a single sequence, rising to 77% with alignment data. ESMFold2 and its computationally lighter counterpart, ESMFold2-Fast, together offer the best combination of speed and accuracy across protein complex prediction benchmarks.
ESMFold2-Fast predicts a 1024-length protein structure in 9.4 seconds while still outperforming existing models on antibody-antigen folding. Speed at this scale changes what's possible: each candidate design becomes a virtual experiment, and hundreds of thousands can be run in a day. This capacity made it possible to scale prediction to all of protein biology — powering ESM Atlas — and to achieve the binder design results described below.
Accelerating the design of therapeutic proteins
Designing a protein that binds a molecular target with the strength and selectivity that biological function requires is a fundamental problem in biochemistry, and has critical implications for medicine. Classical approaches require complex and high throughput laboratory systems that physically search through hundreds of thousands or millions of possible binders, a bottleneck on design of therapeutic agents. Computational design could make finding binders faster and far easier, opening up entirely new applications for protein design.
Antibodies and antibody-derived molecules are a particularly demanding example of the binder design problem because they use unstructured loops to bind their target. Identifying initial candidate molecules with measurable binding to a target can take weeks to months using established experimental methods; the broader antibody development process, from hit to preclinical candidate, typically spans years.
What changes with ESMFold2 is the speed of the earliest stage of that process: the initial search for protein binders. With ESM, we are making a significant step forward on this problem: antibodies can now be designed computationally, with affinities consistent with therapeutic activity.
To evaluate ESMFold2's design capabilities, we used two formats: minibinders — compact, de novo scaffolds with no predetermined structure — and single-chain variable fragment antibodies (scFvs), which retain an antibody's immunoglobulin framework but require design of the highly variable loops that make direct contact with the target. Both are design challenges; scFvs have proven to be particularly difficult for computational approaches. Critically, ESMFold2 was not trained to design antibodies and was not fine-tuned for any of these targets. The binders emerged from the model's latent understanding of protein biology.
ESMFold2 can design binders across multiple disease-relevant targets. This chart shows the fraction of designed proteins that successfully bound their targets in the lab, and how success rates increased as we scaled up the computational search for candidate designs. Click through each target to see the predicted 3D structures of a subset of validated binders.
We selected five targets central to oncology and immunology: EGFR and PDGFRβ, receptor tyrosine kinases implicated in tumor growth; PD-L1 and CTLA-4, immune checkpoints that cancer cells exploit to evade immune surveillance; and CD45, a regulator of immune cell signaling.
ESMFold2's design algorithm follows a simple approach, searching through the joint model of sequence and structure for proteins that are predicted to bind the target. There are two stages to the process. First, candidate generation: searching ESMFold2's representational space to produce tens of thousands or more candidate designs, a process that took roughly two days in these experiments. Second, scoring and ranking: using ESMFold2's confidence scores to evaluate candidates for predicted binding affinity and structural stability, which takes less than a day. Both stages are readily parallelizable.
To test whether more computation improves outcomes, we generated candidate pools at two inference-compute scales and screened the top 84 designs for each target and format at each scale. Averaged across targets, minibinder success rates rose from 54% to 70% at higher compute; for scFvs, success rates nearly doubled, from 12% to 21%. Computational scaling directly translates into better experimental outcomes.
The top-ranked binders were expressed, purified, and characterized in biophysical and cell-based assays. Validated binders exhibited nanomolar binding affinity, specificity, and stability — properties critical for therapeutic relevance — and showed minimal similarity to sequences in public databases. This suggests the model is generating, not retrieving, solutions.

A designed PD-L1-binding scFv engages its target with high affinity and cell-surface specificity, and exhibits functional activity. Top left: ESMFold2-predicted structure of a designed scFv bound to PD-L1, with the modeled interface highlighted in orange. Top middle: biolayer interferometry confirms high-affinity binding by the designed scFv, with a measured KD of 4.3 nM. Top right: in a cell-based PD-1/PD-L1 blockade assay, the designed scFv blocks PD-L1 suppression of T-cell signaling with nanomolar potency; an ESMFold2-designed PD-L1-binding minibinder and negative control are shown for comparison. Bottom: live-cell immunofluorescence shows target-specific binding by the designed scFv to HEK293T cells expressing PD-L1, with nuclei in cyan, target-expressing cells in green, scFv staining in magenta, and the merged image shown at right. Scale bar, 20 µm.
PD-L1 illustrates how this process can design proteins that perform specific biological functions. PD-L1 is expressed by cancer cells to suppress T-cell anti-tumor activity; blocking this suppression is the mechanism of several approved checkpoint inhibitor therapies. An ESMFold2-designed, de novo scFv bound PD-L1 with a measured affinity of 4.3 nM, and in cell-based assays it relieved PD-1/PD-L1-mediated suppression of T-cell signaling with therapeutically relevant nanomolar potency.
These results demonstrate that the earliest stage of binder discovery can be done computationally in days. Research groups working on rare diseases, cancers defined by specific molecular profiles, or targets that have historically attracted little therapeutic attention now have access to these tools.
Although binding affinity can show that a protein design worked in the lab, it doesn't reveal the atomic-level structures underlying function. For a therapeutic protein, the geometry of binding is as important as its strength. EGFR, the human epidermal growth factor receptor, sits on the surface of cells and acts as an antenna for growth signals; malfunctioning forms drive excessive cell proliferation and EGFR is altered in roughly 15% of non-small cell lung cancer cases.
To confirm that ESMFold2's predictions were correct at a structural level, we used cryo-electron microscopy to directly visualize an ESMFold2-designed minibinder bound to EGFR. In this method, protein molecules are illuminated by electron beams, which are able to resolve fine structural features. Rapid sample freezing preserves the proteins in harsh imaging conditions, and computational averaging of thousands of individual images then reconstructs the three-dimensional result.
The microscopy results show that the model predicted the precise location and orientation in which the designed protein bound to EGFR. By directly visualizing the designed binder on its target, we can confirm not only that the model worked, but that it worked for the right reason, closing the loop between AI-generated designs and biological truth.
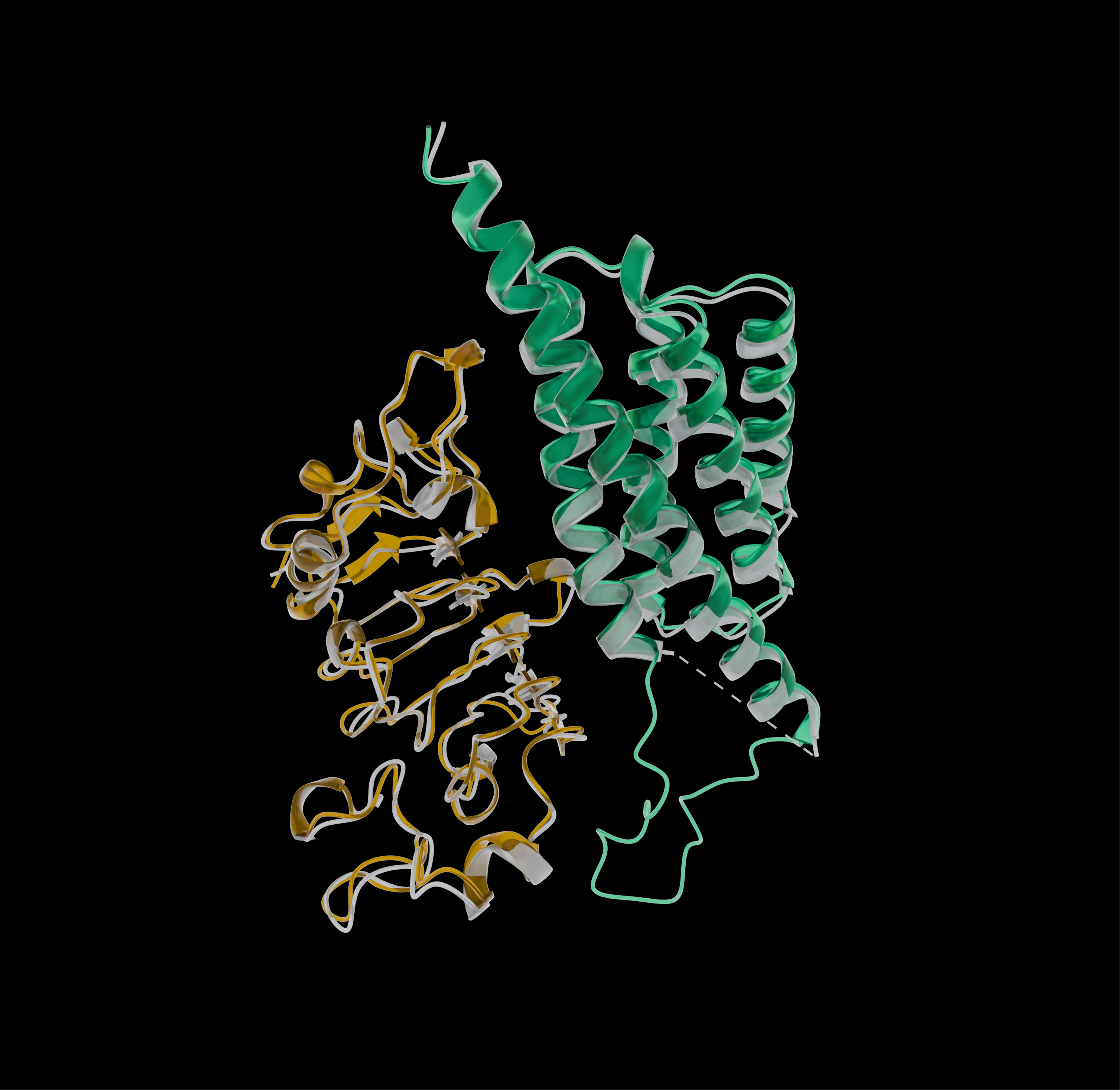
Using cryo-electron microscopy, we verified the structure of an ESMFold2-designed minibinder bound to its target. Here, in green, a designed minibinder binds to human epidermal growth factor receptor (EGFR, yellow) in a computationally predicted model. This predicted model is overlaid with the experimentally observed structure shown in grey. The high concordance between these two models (1.204 Å RMSD between computational and observed minibinder) exemplifies ESMFold2's abilities to precisely design binders and their tight molecular interactions with targets.
The organization of concepts in ESMC's latent space
The principle of reductionism — the idea that it is possible to understand the complexity of biological systems by breaking them into their fundamental components — is one of the central principles of modern biology.
To probe what ESMC has learned, we used sparse autoencoders (SAEs), a technique developed to identify interpretable structure in large language models, to decompose its internal representations into more than 16,000 distinct features.
What we found is that the model, trained only to predict amino acid sequences, with no prior biological knowledge, has independently recovered many of the basic organizing principles of biology, from chemistry of individual amino acids, to local structural interactions, to abstract functional concepts that recur across unrelated proteins, to evolutionary themes connecting all of life.
Just as the same words can be rearranged to create new sentences, features representing fundamental concepts in proteins combine to create complex functions. These features range in specificity from (left) general concepts of structure and biochemistry found across all of biology, to (middle) conserved properties shared across related families, to (right) specific motifs linked to particular functional chemistry. Toggle one, two, or all three features to narrow the group of proteins that correspond with each feature combination. A description of each combination appears in the top right.
At the most fundamental level, SAE analysis shows that ESMC captures amino acid biochemistry. Some features activate on specific amino acids; others represent classes of amino acids (aromatics, small hydrophobics), or context-dependent configurations (lysine residues in low-complexity disordered regions; small side chains embedded in alpha helices).
Beyond residue-level chemistry, the model has learned local interactions that stabilize protein structures and define their key functional regions, including features representing alpha helices and beta sheets, the two primary secondary structure arrangements that form when a protein backbone folds into its most stable conformations.
Such features may be expected in a model that can predict 3D structure with high fidelity. What is striking is that, without any explicit training, the model has identified generalizable concepts — the same ones evolution has independently discovered, across unrelated proteins.
Take, for example, the nucleophilic elbow, a catalytic motif that has evolved independently many times, appearing in enzymes that share no common ancestor and adopt entirely different folds. A single SAE feature activates on this motif across 75 out of 99 relevant enzymes. ESMC was never told what a nucleophilic elbow is. Trained only to predict amino acid sequences, it converged on the same abstraction biochemists use, recovering an underlying rule that evolutionary selection has independently favored across 25 distinct protein folds.
Many features also capture abstract concepts: cellular localization signals, post-translational modifications, local biochemical and electrostatic environments, and functional classes independent of fold. Features even reflect intermolecular interactions — conserved interaction surfaces, short linear motifs, subunit interfaces — inferred from co-evolutionary signals embedded in individual sequences, without ever seeing two proteins at once.
The features learned by ESMC give its representation space an important property: it provides an unbiased lens for connecting proteins whose biological relationship is not apparent from sequence alone. The vast majority of proteins the model was trained on have unknown biology: we don't know what they do, what they interact with, or how they relate to other proteins. But the model has learned features representing them, and the organization of its representation space creates a bridge between known and unknown biology.
ESM Atlas: A map of known and unknown biology
To make this organization of protein biology navigable, we built ESM Atlas, a map of 6.8 billion sequences and 1.1 billion predicted structures. The atlas enables the sequences and structures of proteins across all of life to be studied as a complete picture.
A vivid illustration of what the Atlas can reveal comes from CRISPR-Cas systems, naturally occurring gene-editing systems that bacteria evolved to defend themselves against viruses. The discovery that they could be programmed to edit any DNA sequence was a landmark for biology, enabling high-throughput disease modeling and the development of promising gene therapies. Evolution has produced remarkable diversity in these systems, and the discovery of new variants continues to shed light on their evolutionary origins and to yield editors with improved properties.
In the SAE feature clusters derived from ESM Atlas, a cluster of RNA-guided DNA endonucleases — the functional class that includes CRISPR-Cas systems — brings together eukaryotic Fanzor proteins and their evolutionary ancestor, prokaryotic TnpB, despite their high evolutionary divergence and low sequence similarity.
The cluster includes nine yeast Fanzors identified by Jiang et al., 2023, grouped alongside TnpBs in a way that reflects their shared function. ESMC recovered this evolutionary and functional relationship without being told anything about gene editing or evolutionary ancestry. SAE features describe the functional similarities and differences between these systems, a framework that could help guide the discovery and characterization of new gene-editing tools.
The same unbiased view that reveals these known connections can be applied to the far larger portion of the protein universe where the biology remains unknown.
What this makes possible
The results shared here emerged not from specialized training for a particular target, but from a model of protein biology that can now be searched computationally.
Computing structures for 1.1 billion proteins in a week, or screening millions of candidates in hours, isn't just faster science. The ability to run experiments virtually, at a scale that isn't possible in the laboratory, starts to unlock new possibilities in biology.
While disease follows common patterns, much of disease is individual, and for certain diseases like cancer and rare disease the potential is most immediate. We have shown that ESM can design lab-validated protein binders for five clinically relevant targets in days. What this work changes is the speed of the earliest stage of that path, made possible because these tools are now available to any researcher, anywhere, who wants to use them.
The future of biology will depend on systems that combine scaled data, predictive models, and experimental feedback. This world model is an important step toward that future: an open foundation for understanding proteins, discovering new biology, and designing molecular tools that can help prevent, treat, or cure disease.
Availability and access
ESMFold2, ESMC, and ESM Atlas are available today at the Biohub Platform. The full results are described in our preprint, which will be posted on bioRxiv. All models have been released under an MIT license.
Biohub conducts proactive assessment of benefits and potential risks of our models, tools and datasets prior to release, and implements guardrails where necessary. We actively engage with the scientific community, stakeholders and domain experts to advance innovation as well as best practices for responsible development. Risk assessment was conducted for each of the components of this release, including our ESMC Cambrian models, ESMFold2, ESMC SAEs, ESM Atlas, and binder design system.
We are partnering with a number of platforms to make the models available:
- AWS Bio Discovery
- Benchling AI
- Modal
- Phylo
- SandboxAQ
- Tamarind Bio
- Tool Universe
NVIDIA's TransformerEngine and cuEquivariance kernels were used in the training of ESMC and ESMFold2. In collaboration with NVIDIA, we have also incorporated optimized kernels for context-parallel and accelerated inference into the open-source ESMFold2 models.